
The following is a list of research projects I have contributed to.
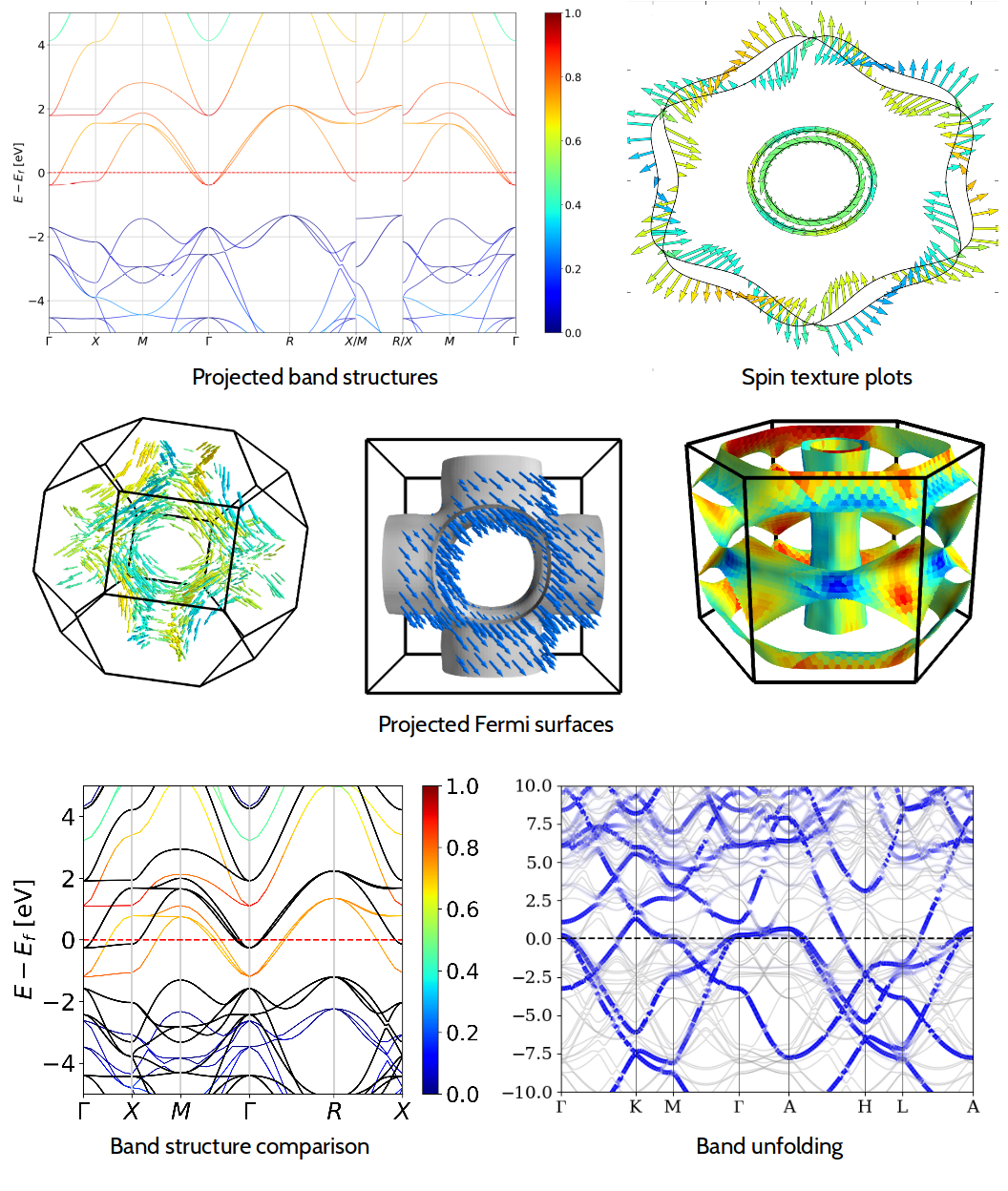
PyProcar
A Python library for electronic structure pre/post-processing
PyProcar is a robust, open-source Python library used for pre- and post-processing of the electronic structure data coming from DFT calculations. It provides a set of functions that manage data obtained from the PROCAR format. Basically, the PROCAR format is a projection of the Kohn-Sham states over atomic orbitals. That projection is performed to every k-point in the considered mesh, every energy band and every atom. PyProcar is capable of performing a multitude of tasks including plotting plain and spin/atom/orbital projected band structures and Fermi surfaces- both in 2D and 3D, Fermi velocity plots, unfolding bands of a super cell, comparing band structures from multiple DFT calculations, plotting partial density of states, filtering bands and generating a k-path for a given crystal structure. Currently supports VASP, Elk, Quantum Espresso, Abinit, Lobster, Siesta and DFTB+.
Reference:
1. Lang, L., Tavadze, P., Tellez, A., Bousquet, E., Xu, H., Muñoz, F., Vasquez, N., Herath, U., & Romero, A. H. Expanding PyProcar for new features, maintainability, and reliability. Computer Physics Communications 297, 109063 (2024). DOI: https://doi.org/10.1016/j.cpc.2023.109063
2. Herath, U., Tavadze, P., He, X., Bousquet, E., Singh, S., Muñoz, F., & Romero, A. H. PyProcar: A Python library for electronic structure pre/post‑processing. Computer Physics Communications 251, 107080 (2020). DOI: https://doi.org/10.1016/j.cpc.2019.107080

DMFTwDFT
An open-source code combining Dynamical Mean Field Theory with various Density Functional Theory packages to study strongly correlated materials
DMFTwDFT is an open-source, user-friendly framework developed in Python, Fortran and C/C++, combining DMFT with various DFT codes interfaced through the Wannier90 package. It allows users to perform full charge self-consistent DFT+DMFT calculations using minimal commands followed by post-processing functions including DMFT self-energy, total energy, band structures, projected DOS, and effective mass enhancements. DMFTwDFT uses Wannier orbitals for constructing the hybridization and correlation subspaces to perform DMFT loops by taking advantage of the Wannier90 interface between various DFT codes. It also equips a library mode to link the module for computing a DMFT density matrix and updating a charge density within the DFT loops allowing implementing full charge self-consistency within various DFT codes without modifying their source codes significantly. Currently supports VASP, Quantum Espresso (one-shot DMFT), and Siesta (one-shot DMFT).
Reference:
Singh, V., Herath, U., Wah, B., Liao, X., Romero, A. H., & Park, H. DMFTwDFT: An open‑source code combining Dynamical Mean Field Theory with various density functional theory packages. Computer Physics Communications 261, 107778 (2021). DOI: https://doi.org/10.1016/j.cpc.2020.107778

MechElastic
A Python Library for Analysis of Mechanical and Elastic Properties of Bulk and 2D Materials
MechElastic allows users to calculate several important physical properties such as elastic moduli, melting temperature, Debye temperature, elastic wave velocities, elastic anisotropy, etc. for all crystalline 2D and 3D systems using output data from an elastic tensor calculation. It can also be used to test the mechanical stability of any bulk system. Additionally, MechElastic allows performing Equation of State (EOS) analysis provided inputs of Volume and Energy or Volume and Pressure and automatically provides information on phase transitions. Currently supports VASP, Abinit and Quantum Espresso.
Reference:
Singh, S., Lang, L., Dovale‑Farelo, V., Herath, U., Tavadze, P., Coudert, F.‑X., & Romero, A. H. MechElastic: A Python library for analysis of mechanical and elastic properties of bulk and 2D materials. Computer Physics Communications 267, 108068 (2021). DOI: https://doi.org/10.1016/j.cpc.2021.108068

FHI-aims
An all-electron electronic structure theory with numeric atom-centered orbitals
FHI-aims is an all-electron electronic structure code based on numeric atom-centered orbitals. It enables first-principles simulations with very high numerical accuracy for production calculations, with excellent scalability up to very large system sizes (thousands of atoms) and up to very large, massively parallel supercomputers (ten thousand CPU cores).
Reference:
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter, and M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Computer Physics Communications 180, 2175 (2009). DOI: https://doi.org/10.1016/j.cpc.2009.06.022

ELSI (ELectronic Structure Infrastructure)
A unified software interface designed for electronic structure codes to connect with various high-performance eigensolvers and density matrix solvers
ELSI provides and enhances scalable, open-source software library solutions for electronic structure calculations in materials science, condensed matter physics, chemistry, and many other fields. ELSI focuses on methods that solve or circumvent eigenvalue problems in electronic structure theory. The ELSI infrastructure should also be useful for other challenging eigenvalue problems.
Reference:
1. V. W. Yu et al., ELSI — An open infrastructure for electronic structure solvers, Computer Physics Communications 256, 107459 (2020). DOI: https://doi.org/10.1016/j.cpc.2020.107459
2. V. W. Yu et al., ELSI: A unified software interface for Kohn–Sham electronic structure solvers, Computer Physics Communications 222, 267 (2018). DOI: https://doi.org/10.1016/j.cpc.2017.09.007

HybriD3
A curatable database for experimental and theoretical data on hybrid organic-inorganic materials
Hybrid organic-inorganic materials offer a unique opportunity for the discovery and refinement of new functional semiconductor materials with fine-tuned properties, controlled at the atomic scale by organic chemistry and organic-inorganic synthesis and processing. The HybriD$^3$ project accelerates the Design, Discovery and Dissemination (D3) of new crystalline organic-inorganic hybrid semiconductors in a collaborative effort between a consortium of researchers in the NC Triangle area and beyond. It is based on the MatD$^3$ framework written in Python-Django and uses SQL for database functions.
Reference:
R. Laasner, X. Du, A. Tanikanti, C. Clayton, M. Govoni, G. Galli, M. Ropo, and V. Blum, MatD$^3$: A Database and Online Presentation Package for Research Data Supporting Materials Discovery, Design, and Dissemination, JOSS 5, 1945 (2020). DOI: https://doi.org/10.21105/joss.01945

Springer Materials
The research solution for identifying material properties
Springer Materials provides curated data and advanced functionalities to support research in materials science, physics, chemistry, engineering, and other related fields.
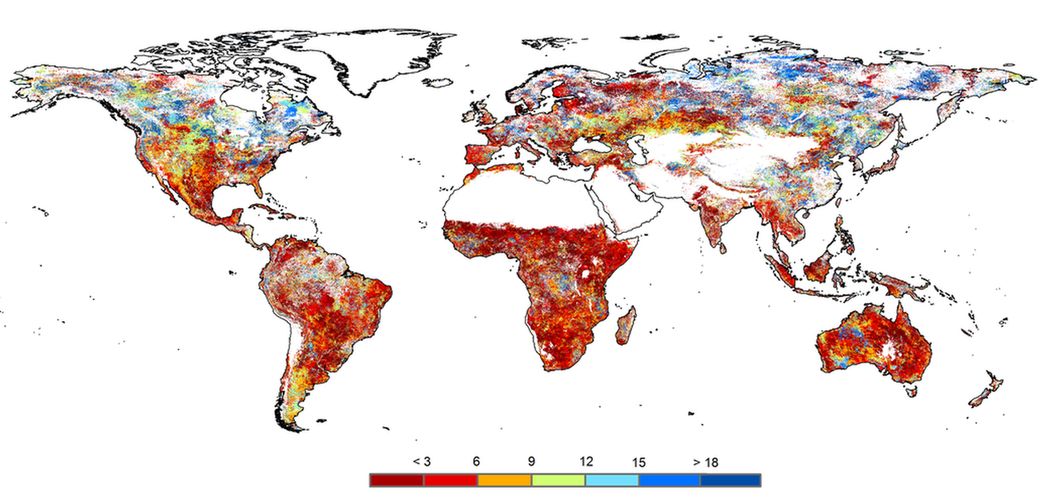
convertSPEI
This program converts time series Standardised Precipitation-Evapotranspiration Index (SPEI) data from the netcdf format to a csv format which could be read by a program such as Excel. It is parallelized through Python’s multiprocessing library. The SPEI data is available at https://spei.csic.es/index.html.
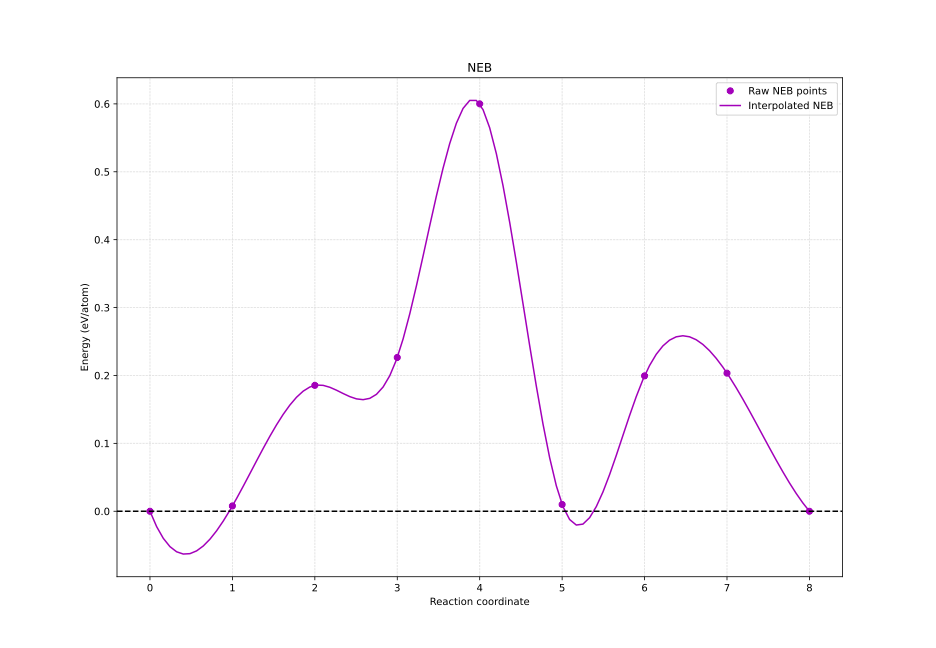
NEBgen
NEBgen prepares POSCAR’s for Nudged Elastic Band (NEB) calculations through Distortion Symmetry Method with VTST and DiSPy. The nudged elastic band (NEB) method is a popular method for calculating the minimum energy pathways of kinetic processes. Although, linear interpolation between an initial and final structure (image) provides a decent estimate for the minimum energy pathway, the Distortion Symmetry Method takes into account symmetry-adapted perturbations to systematically lower the initial path symmetry, enabling the exploration of other low-energy pathways that may exist.
PyChemia
PyChemia is an open-source Python library for materials structural search providing an agnostic framework for materials discovery and design using a variety of methods from Minima Hoping to Soft-computing based methods. PyChemia is also a library for data-mining, using several methods to discover interesting candidates among the materials already processed. Currently supports VASP, ABINIT, Octopus, DFTB+, and Fireball.
mdwc3
The molecular dynamics with constraints (mdwc3) package is a command line open-source Python program for molecular dynamics simulations. It performs constraint MD simulations with either NPT (keeping pressure constant with the Parrinello-Rahman Lagrangian, and keeping the temperature constant with the Nosé-Hoover thermostat) or NVT (keeping the temperature constant with the Nosé-Hoover thermostat). mdwc3 performs constraint MD simulations following the SHAKE algorithm and allows constraints for bond distances, angles, atomic positions, lattice parameters (a, b, c), angles between lattice vectors, and volume of the unit cell.